The post EMA Issues Guidance on Risk Minimisation Measures for Medicinal Products with Embryo-Foetal Risks appeared first on UBC.
]]>This Addendum to Module XVI provides essential guidance on when and how to implement risk minimisation measures (RMMs) for products with embryo-foetal risks while ensuring patient medical needs are not compromised when no suitable alternative treatments are available.
The Addendum offers some clarity as to what constitutes a Pregnancy Prevention Programme (PPP). Historically, without clear guidance, there has been a tendency to equate a PPP with a Controlled Access Programme such that specific activities, e.g. pregnancy testing must be performed, documented and verified prior to dispensing the medicinal product. This has proved challenging in several European Member States where either local regulations, healthcare organization, or culture have created obstacles to such implementation.
GVP Module XVI Revision 31 introduced concept of Control Tools and listed a number of different activities for consideration. These included one or more of the following:
- Healthcare professional qualification
- Healthcare facility accreditation
- A traceability system
- A system for documenting exchange of information
- Certificates of medical interventions which are required for the prescribing or dispensing of the medicinal product
Addendum I, released in August 2025,2 describes intended actions for risk minimisation applied to embryo-foetal risks (e.g., counselling, taking actions to avoid pregnancy, pregnancy testing, supervision of treatment and avoiding blood or sperm donation if applicable) and the tools that can be used as RMMs applied to embryo-foetal risks. These tools include routine RMMs such as:
- Product labelling
- Visual enhancements, special warnings on packaging
- Disallowing free samples
Additional RMMs include:
- Educational materials for healthcare professionals
- Educational materials for patients
Addendum I further states in exceptional situations of embryo-foetal risks of a medicinal product, intended actions for risk minimisation and RMM tools can be combined to form a PPP. It states that a PPP is constituted by at least the following:
- Contraindication or a contraindication unless there is no suitable alternative treatment during pregnancy
- Risk counselling
- Taking actions to avoid pregnancy
- Pregnancy testing
- Supervision of treatment
- Reminder statements of risk of embryo-foetal risk on outer packaging
- Educational material for healthcare professionals
- Educational material for patients
The Addendum goes on to say that the PPP may include one or more of the control tools listed above but not to the extent that a control tool is a mandatory part of a PPP.
UBC welcomes this guidance which provides clarity on what constitutes a PPP and believes it is key to ensuring patient access to medicines across all Member States in the EU while maintaining patient safety.
This clarification related to EU risk minimization measures comes at a time where risk management shifts have also been seen in the United States. In July 2025, FDA announced that risk evaluation and mitigation strategy (REMS) requirements for embryofetal toxicity risks would be removed from all endothelin receptor antagonist medicines.3 The REMS requirements had previously been based on findings from animal studies and FDA monitoring of data over time did not show a pattern of congenital malformations consistent with what was observed in animal embryofetal toxicity studies.
This evolution in risk management, grounded in evidenced-based decision making, is a promising step to ensure patients have access to life-changing medicines in an ever-changing healthcare landscape.
References
1. European Medicines Agency. Guideline on good pharmacovigilance practices (GVP). Module XVI – Risk minimisation measures (Rev 3). July 26, 2024. Accessed September 16, 2025. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guideline-good-pharmacovigilance-practices-gvp-module-xvi-risk-minimisation-measures-rev-3_en.pdf
2. European Medicines Agency. Guideline on good pharmacovigilance practices (GVP). Module XVI Addendum I – Risk minimisation measures for medicinal products with embryo-fetal risks. August 22, 2025. Accessed September 16, 2025. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-xvi-addendum-i-risk-minimisation-measures-medicinal-products-embryo-fetal-risks_en.pdf
3. U.S. Food & Drug Administration. Endothelin Receptor Antagonist REMS Information. July 3, 2025. Accessed September 16, 2025. https://www.fda.gov/drugs/information-drug-class/endothelin-receptor-antagonist-rems-information
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.
UBC is a leader in risk management and REMS activities and can help Sponsors develop, implement, and assess risk management strategies. Our consultation, design, implementation, and evaluation offer a cohesive experience throughout a product’s life cycle. Our subject matter experts (SMEs) remain on the cutting edge of developments across all facets of risk management and can support customers in securing safe and appropriate use of their products.

About the Author
Dr. Janine Collins, MBBS, LLM. Vice President, Medical, Scientific & Real-World Research
Dr. Janine Collins, MBBS, LLM, is Vice President, Medical, Scientific & Real-World Research at UBC. She provides leadership, direction, and guidance supporting customers in the creation of core Risk Management Plans and global and local strategy for Risk Management activities. Her experience spans a wealth of therapeutic areas and latterly has focused on several rare diseases in both adults and pediatrics including metabolic disorders, inborn errors of metabolism, neurodegenerative disorders, and pediatric and adult oncology.
The post EMA Issues Guidance on Risk Minimisation Measures for Medicinal Products with Embryo-Foetal Risks appeared first on UBC.
]]>The post To Rescue or Not to Rescue Your Study: How to Spot Trigger Points in Strategic Partnerships appeared first on UBC.
]]>Which makes it even more complicated when you realize a mid-study change might be necessary. Switching service providers in the middle of a clinical trial is never simple. Patient safety, accountability, personal relationships, and significant financial investment are all on the line. With RFP-to-contract cycles stretching from three to 12 months, the sunk costs in time and effort can feel overwhelming.
Even with these pressures, a moment comes when the risks to data, timelines, and delivery are too great, and the same conclusion is drawn: it’s time to move on. Some signals are obvious, while others are much quieter. At UBC, our rescue experts have seen them all. Here are a few of the most common — and what to do about them.
Trigger Point 1: Communication Worries
You’re nearing a study first: site activated, patient screened, IP delivery, but responsiveness on the CRO side has stalled. Your repeated outreaches go unanswered, and it’s unclear who owns communication and accountability. Every email gets forwarded to a new name in the reply chain. Every meeting has a new project management person. You’re spending twice the time you budgeted just to get answers to basic questions about your study.
This is one of the most frequent pain points Sponsors cite, particularly with large CROs. They highlight shifting teams, the breakdown of communication, spotty governance, and lack of personalized attention as their biggest frustrations. Yet even though these communication breakdowns are painful, they are often reversible. The right CRO will respond when feedback is delivered clearly, when additional transparency is requested, and when expectations are tied to measurable outcomes.
“We promise good communication” is not a tagline; it’s a mandate. It demands a competent team, a well-established structure, and an organization devoted to study ownership and strong training.
Our Approach:
At UBC, communication isn’t left to chance. We apply a centralized model, assigning a single point of contact who serves as the fulcrum for study management. In a rescue, that focus becomes even sharper, aligning our expert Rescue Team with specialized staff that can anticipate bottlenecks, communicate effectively, and neutralize transition risk.
“We think in terms of creating a tailored clinical support team. This isn’t a call center or an inbox—these are real people dedicated to the transition, one single point of contact,” Rahul Malhotra, Program Director explains. “Sometimes the sponsor is just overwhelmed, and it’s our job to show up consistently and provide that stability.”
Trigger Point 2: The Bait and Switch
This one is infamous. “It feels like every conference I’ve been to this year,” notes JB Flinders, Executive Director of Strategic Client Engagement, “Sponsors commiserate about how their CRO’s ‘A team’ was replaced by the ‘C team’, rarely with an explanation why.” It’s a pain many sponsors feel. You sign with a CRO expecting a specific team, a defined price, and a clear delivery path. Then post-award, things start to shift. The team changes, timelines extend, and your inbox is suddenly full of change order requests. The CRO you thought you hired no longer resembles the one now running your trial.
In the highly competitive environment of drug development, this can be disastrous. While the most evident costs are budgetary, more subtle costs in credibility, potential market share, and stakeholder relationships begin to emerge. Your “strategic partner” suddenly feels less integrated and more transactional. The essence of partnership may still be there, but the real test is what happens when things go wrong.
Our Approach:
No two rescues are alike, but one truth remains constant: recovery depends on readiness. Our experienced Rescue Team brings decades of experience managing complex, at-risk trials across numerous therapeutic areas. We have encountered every rescue challenge imaginable and have solutions ready from Day 1.
We know that rescue work is as much about rebuilding trust and stabilizing the relationship – with you, your sites, and your patients – as it is about operational turnaround. That process begins long before contract signature, building proposals grounded in realism with an eye toward sustainable, measurable, and dependable results.
Counter Point: Radical Accountability
A successful partnership isn’t about hitting the mark 100% of the time, it’s about trust and accountability. In clinical development, failure often teaches faster than success, and your most valuable partners are those who own their mistakes, improve processes, and move forward without any loss in momentum.
For the studies you have, it may be worth asking: Do you enjoy working with your project team? Is senior leadership actively engaged or CC’d occasionally in a long chain of emails? Do you have transparency into budgets, study team decisions, and milestones? These answers may be strong indicators of long-term compatibility with your CRO partner.
Do you enjoy working with your project team? Is senior leadership actively engaged or CC’d occasionally in a long chain of emails? Do you have transparency into budgets, study team decisions, and milestones?
Ultimately, while our endpoints are data-focused, we are an industry of relationships. Each of us is a steward, working towards a singular goal of better outcomes for patients. Success isn’t one-size-fits-all, but strong communication, meaningful connections, and collaborative and transparent processes ensure that positive outcomes are more likely to be met.
Final Thought: What the Data Says
A recent Tufts Center for the Study of Drug Development analysis confirms growing sponsor concern about the operational stability of large CROs. It notes performance volatility and turnover as primary risks, which reinforces the need for true partnership models rather than traditional transactional outsourcing. In an era defined by volatility, UBC is here to support our Sponsors by pairing operational excellence, data-driven strategies, dedicated oversight, and an environment of trust and transparency.
Read part one of our series here.
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.

About the Author
JB Flinders, Executive Director, Strategic Client Engagement
With over 16 years of clinical and academic research experience, JB Flinders has an extensive track record of driving successful operational, feasibility, and analytic strategy across the full scope of therapeutic areas and trials including multiple rescue studies.

Rahul Malhotra, Project Director, Global Project Delivery
Rahul Malhotra is an experienced clinical research Director with more than 17 years in global project delivery. He is recognized for his strategic oversight, strong client partnerships, and successful delivery of studies Globally, spanning Real-World Evidence and interventional research across a wide range of therapeutic areas.

Natania Barron, Sr. Director, Marketing
Natania Barron is a life sciences marketing professional with over 20 years of experience. Her passion is storytelling at the intersection of data and narrative.
The post To Rescue or Not to Rescue Your Study: How to Spot Trigger Points in Strategic Partnerships appeared first on UBC.
]]>The post The Critical Role of the QPPV: Insights from UBC’s Pharmacovigilance Experts appeared first on UBC.
]]>Q: For companies entering Europe, having a QPPV is a legal requirement. What does this role actually involve, and why is it so critical?
A: The QPPV role was formally introduced in 2012 under European GVP legislation. It’s not just a regulatory box to check – it’s a mandatory, legally defined function central to patient safety.
The QPPV ensures oversight of a product’s safety profile, benefit–risk balance, and all associated pharmacovigilance activities. Put simply: without a QPPV, you cannot legally enter the European market. The role is the cornerstone of ensuring patients are protected and that regulatory expectations are consistently met.
Q: At what stage should companies bring a QPPV on board, and how does the role add value across the product lifecycle?
A: A QPPV should be engaged before submission. Regulatory dossiers require a description of the pharmacovigilance system, and documents like the Risk Management Plan (RMP) and Pharmacovigilance System Master File (PSMF) need QPPV review and approval.
The ideal time is typically toward the end of Phase III clinical trials – when companies begin developing the RMP and assembling their submission package. Early involvement ensures a smoother transition to regulatory approval and post-marketing oversight.
Q: What are the benefits of outsourcing the QPPV function to an experienced global partner like UBC, versus keeping it in-house?
A: Outsourcing provides unmatched breadth of experience, flexibility, and scalability. While in-house roles may offer easier oversight of internal systems, a QPPV at UBC brings knowledge from multiple clients, audits, and inspections across diverse therapeutic areas and geographies.
This cross-company experience helps identify potential pitfalls early, apply best practices, and ensure a resilient pharmacovigilance framework. At UBC, QPPVs are backed by an entire infrastructure of experts – supporting seamless operations, inspection readiness, and global compliance.
Q: UBC has over 30 years of global experience in pharmacovigilance. How does this translate into value for clients?
A: UBC has decades of hands-on inspection and audit experience with regulators across Europe (MHRA in the UK, PEI in Germany, MPA in Sweden, and more) as well as in the US, Japan, and Switzerland.
This global footprint means we understand regulatory nuances across markets and can support clients with cross-continental teams, local expertise, and a quality-driven infrastructure. Our clients benefit from a truly global perspective combined with local market knowledge.
Q: How does the QPPV role integrate with other stakeholders – both internally and externally?
A: The QPPV is not a standalone figure but a hub of communication and oversight. Internally, the QPPV works closely with regulatory affairs, quality, medical affairs, and clinical teams. Externally, the role connects with clients’ own departments as well as health authorities.
Strong communication pathways and structured processes are essential. At UBC, we emphasize robust, transparent, and proactive communication – ensuring alignment across stakeholders while maintaining patient safety as the top priority.
Q: What role does technology play in enabling effective QPPV oversight?
A: Technology is indispensable. Pharmacovigilance systems manage vast volumes of safety data daily, from spontaneous reports to literature monitoring and signal detection.
UBC leverages validated safety databases, advanced signal detection tools, and technology-driven literature review systems. Our approach ensures real-time oversight, audit-ready documentation, and efficient regulatory responses.
Looking ahead, we’re actively exploring AI solutions to further enhance efficiency and compliance, while staying aligned with evolving legislation.
Q: What are the most common pitfalls companies face when establishing QPPV oversight, and how does UBC help avoid them?
A: The biggest issues include:
- Late engagement of the QPPV – often too close to submission deadlines.
- Misunderstanding EU GVP legislation, particularly among companies entering Europe from outside markets.
- Poor communication and coordination across stakeholders.
UBC addresses these challenges by engaging early, applying project management discipline, conducting audits and mock inspections, and ensuring systems are inspection-ready at all times.
The UBC guiding principle: patient safety first, compliance always.
Closing Thoughts
As Peter noted, appointing a QPPV is far more than a regulatory requirement – it’s a commitment to patient safety and compliance excellence.
With UBC’s decades of experience, global inspection track record, and technology-enabled systems, we partner with companies to navigate the complexities of pharmacovigilance with confidence.
Listen to the interview below:
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.
The post The Critical Role of the QPPV: Insights from UBC’s Pharmacovigilance Experts appeared first on UBC.
]]>The post Evidence Matters: Joel White of Marketcap Consultants appeared first on UBC.
]]>The post Evidence Matters: Joel White of Marketcap Consultants appeared first on UBC.
]]>The post Rethinking Decentralization in Direct-to-Patient Studies: A Conversation with Aaron Berger appeared first on UBC.
]]>Listen to the full interview to hear Aaron Berger share how UBC is pioneering decentralized clinical trial models that prioritize patient access, engagement, and data integrity. He discusses the operational challenges and opportunities in shifting from traditional site-based studies to direct-to-patient approaches, and how UBC’s Evidence Development Solutions are uniquely positioned to support sponsors through this transformation.
Berger also highlights the importance of aligning technology, regulatory standards, and patient needs to create trials that are not only feasible but impactful. This episode offers a compelling look at the future of clinical research and the strategic role UBC plays in driving innovation across the product lifecycle.
Why Decentralized Clinical Trials Matter
Decentralized clinical trials (DCTs) are designed to reduce patient burden and increase accessibility by enabling participation from home or local settings. Berger emphasizes that DTP models are not just operationally efficient—they’re essential for improving diversity, retention, and real-world relevance.
Scientific and Operational Benefits of DTP Models
UBC’s approach to evidence development integrates decentralized strategies to:
- Enhance patient recruitment and retention by removing geographic and logistical barriers
- Improve data quality through real-world data collection and remote monitoring
- Support regulatory compliance while maintaining scientific rigor
- Enable flexible study designs that adapt to patient needs and sponsor goals
These benefits align with UBC’s commitment to delivering fit-for-purpose evidence that supports product development, regulatory approval, and market access.
Technology-Enabled Patient Experiences
Technology is a key enabler of DTP studies. Berger discusses how tools like eConsent, telehealth, mobile sample collection, and digital patient-reported outcomes (ePROs) are streamlining the research process while maintaining high standards of data integrity.
UBC ensures that technology is implemented with a focus on usability, accessibility, and compliance, helping sponsors meet patients where they are—physically and digitally.
UBC’s Evidence Development Solutions: Driving Innovation
UBC’s Evidence Development Solutions are built to support sponsors across the product lifecycle—from early-phase studies to post-marketing evidence generation. Berger highlights how UBC combines scientific expertise, operational excellence, and patient-first thinking to deliver impactful results.
Whether through hybrid or fully decentralized models, UBC helps sponsors generate regulatory-grade, real-world evidence that informs decision-making and improves patient outcomes.
Looking Ahead: The Future of Patient-Centric Research
As the industry continues to embrace decentralization, the focus is shifting from feasibility to optimization. Berger encourages stakeholders to prioritize trust, communication, and empathy—the human elements that make DTP studies successful.
UBC remains committed to advancing decentralized research models that are scientifically sound, operationally feasible, and deeply aligned with patient needs.
Ready to Rethink Your Evidence Strategy? Let’s Talk.
Decentralized clinical trials are more than a trend—they’re a strategic imperative for sponsors seeking to generate high-quality, patient-centric evidence. At UBC, we combine scientific rigor, operational excellence, and deep patient engagement to help you design and execute studies that deliver meaningful results.
Whether you’re exploring hybrid models, fully remote trials, or real-world evidence strategies, our team is here to guide you every step of the way.
Connect with us today to learn how UBC’s Evidence Development Solutions can support your next study and accelerate your path to market.
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.
The post Rethinking Decentralization in Direct-to-Patient Studies: A Conversation with Aaron Berger appeared first on UBC.
]]>The post Beyond Enrollment Woes: Why Sponsors Seek Rescue in Tumultuous Times appeared first on UBC.
]]>In concert with that statistic, however, is the growing trend of rescue studies. The two are invariably intertwined—disappointed sponsors look to tighten timelines when studies just can’t get off the ground, which often means switching their service partners. Data is relatively scarce on specifics—as what determines a “rescue study” differs from company to company. Global Data’s statistics for 2018 cites about 20% of trials that year being rescued, but there is no current standard for measurement, and no public up-to-date statistics. Either way, rescues are here to stay.
Lagging enrollment is just part of the struggle for sponsors, and likely an easy scapegoat for a far more complex problem. In fact, in many cases, sponsors are looking for rescues due to factors dragging down or putting their trials at risk that have nothing to do with enrollment.
Lagging enrollment is just part of the struggle for sponsors, and likely an easy scapegoat for a far more complex problem.
Rahul Malhotra, Project Director, Evidence Development Solutions at UBC, has observed a surprising shift. “You’re seeing studies now with all sites activated and patients enrolling, but dissatisfaction and distrust on the sponsor side. Sometimes it’s due to having too many vendors in the mix; other times it’s a data quality issue. And often, it’s full-scale lack of communication and coordination.”
Rahul further explains that some sponsors may feel as if they can’t afford to rescue, given the perceived financial costs in developing such a transition. Adding another vendor into the mix is overwhelming, especially later in the game, because it doesn’t just mean resetting the clock on an already delayed or at-risk program, but investing in a transition and rebuilding trust without data loss or change in momentum.
However, in some cases, it’s too risky and too expensive to remain in the same partnership, so sponsors gravitate toward solutions providers who can be flexible and strategic, experienced in the nuanced shift and lift needed to transition successfully.
Although there are dozens of technical aspects of this handover, one thing that most often is overlooked—and hardest to maintain—is communication. Though the word has become more of a corporate buzzword these days, in rescue studies there is no room for compromise. New team staff must maintain consistent, clear, and direct communication during handoff, and sponsors need access and transparency throughout.
“Full program management requires a seamless experience, and communication is at the heart of that,” Rahul says. “It’s about partnering with a team who can pre-emptively anticipate what needs to happen, make those changes, and keep the program moving, so sponsors can get back to focusing on what matters”.

These solutions allowed timely approval from regulatory authorities and study start-up. Our rescue team achieved a first site initiation visit only three months after CRO handover, and the first subject identified was dosed one month later.
Patient screening and enrollment was four months ahead of schedule in the first year. The six-month goal of screened patients was 41, and the trial was able to screen over 200 patients during that time. The trial’s focus on diverse recruitment led to a more comprehensive understanding of the treatment’s potential benefits and risks across various populations.
Comprehensive trial design and planning resulted in zero inclusion/exclusion protocol amendments. This was a key factor in minimizing disruptions to trial progress, proving study and data integrity to patients and investigators, and allowing enrollment to stay ahead of schedule.
Implementation of modernized study design kept the rescue in lock step with established startup, recruitment, treatment, and follow-up timelines. At the time of publication, the trial was trending for an early completion, which reduces costs without compromising research effectiveness and integrity.
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.
About the Author
JB Flinders, Executive Director, Strategic Client Engagement
With over 16 years of clinical and academic research experience, JB Flinders has an extensive track record of driving successful operational, feasibility, and analytic strategy across the full scope of therapeutic areas and trials including multiple rescue studies.
Rahul Malhotra, Project Director, Global Project Delivery
Rahul Malhotra is an experienced clinical research Director with more than 17 years in global project delivery. He is recognized for his strategic oversight, strong client partnerships, and successful delivery of studies Globally, spanning Real-World Evidence and interventional research across a wide range of therapeutic areas.
Natania Barron, Sr. Director, Marketing
Natania Barron is a life sciences marketing professional with over 20 years of experience. Her passion is storytelling at the intersection of data and narrative.
The post Beyond Enrollment Woes: Why Sponsors Seek Rescue in Tumultuous Times appeared first on UBC.
]]>The post Regulatory Submissions with Real-World Evidence: Highlights from the Joint Duke Margolis Institute and FDA Public Meeting appeared first on UBC.
]]>The Advancing RWE program was established in 2022 with three goals:
- Identify approaches for generating RWE that meet regulatory requirements in support of labeling for effectiveness
- Develop agency processes that promote consistent decision-making and shared learning regarding RWE
- Promote awareness of characteristics of RWE that can support regulatory decisions by allowing FDA to discuss study designs considered in the Advancing RWE Program in a public forum.
On September 23rd, the Duke Margolis Institute for Health Policy convened a public meeting in conjunction with the US FDA on the topic of Regulatory Submissions with Real-World Evidence (RWE): Successes, Challenges, and Lessons Learned, with over 4,000 virtual and in-person attendees. This meeting completes the FDA’s 2025 RWE-PDUFA VII commitments for the Advancing RWE program and included case studies with a focus on how to generate RWE for regulatory requirements. By the end of 2026, the FDA will use lessons learned from their Advancing RWE Program to update existing, or generate new, RWE-related guidance documents.
In opening remarks, Dr. Sarah Brenner, Principal Deputy Commissioner at the FDA, described ongoing efforts in RWE as part of the path toward a “more modern and agile FDA.” Modernization and agility were repeated themes throughout the day.
Another theme was the importance for sponsors to engage with the agency early as a thought partner in study design, to determine the best way to incorporate RWE. According to sponsors presenting case studies, this approach was one of their biggest success factors. Case studies presented largely focused on data source fitness for use, appropriateness of the RWE study design, and regulatory considerations, which is consistent with current guidance.
The panel session focused on the strengths and challenges of using RWD. Panelists were from Office of Medical Policy, CBER, Abbott, Orchard Therapeutics, and Bristol Myers. The panel spoke on a number of topics including data source selection and fitness for use:
- The agency does not endorse any particular data source and is open to considering any data source that meets requirements, including natural history studies, registries, EHRs, claims, hospital charge master data.
- Data feasibility studiesa are required to understand relevance and reliability of the data, data quality, and population fit.
- The data source(s) should contain the outcome or exposure of interest, and outcomes should be objective (e.g., overall survival).
- The data source should have enough patients for the study.
- Robust sensitivity analyses are needed on endpoints to test assumptions on relevance and reliability of the data.
- When there is limited of data, such as with ultrarare diseases, natural historyb cohorts become very important for direct comparisons.
When the topic of broader use of RWE came up, beyond unmet need and into more general diseases, there was tempered optimism among the panel. The field is evolving, and there is some expectation that the use of RWE will become much more common, including greater use in registrational trials,c post-marketing requirements (PMRs), and post-marketing commitments (PMCs). While there is more bias potential in observational studies than in controlled trials, but with larger sample sizes (e.g., in more general diseases), sponsors may be able to design studies better and include more complex analyses to minimize bias. However, it was clear that the continued expectation is that the role of RWE is to complement, not replace, controlled trials.
Sponsors and regulators agreed that they are all invested in seeing greater use of RWE in the future. Accordingly, the agency has new efforts underway to look at methods to promote alignment and consistency. Ultimately, the FDA is looking to lead efforts to advance the field globally.
Additional resources:
- To learn more about data source feasibility assessments, read UBC’s recently posted blog on this topic, titled “Is Your Real-World Data ‘Fit-for-Purpose’? The Critical Role of Feasibility Assessments.”
- To learn more about Natural History Studies, read UBC’s white paper, “Understanding the Natural History of Disease.”
- To learn more about linking real-world data to clinical studies, read UBC’s publications:
- Case study: Enriching clinical studies with longitudinal real-world data
- White paper: Gain Strategic Insights by Linking Clinical Trial Data to Real World Data
- Blog: Longitudinal Real-World Data: How to Gain Deeper Insights Into Your Clinical Studies
- Blog: RWD Enrichment Studies: Linking Pre- and Post-Trial Data Utilizing Tokenization
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.

About the Author
Judy Lytle, PhD, MBEE, PMP. Executive Director, Evidence Development Study Solutions
Judy Lytle serves as the Executive Director of Evidence Development Study Solutions for UBC. Dr. Lytle joined UBC in 2023, bringing more than 15 years of experience in life science and healthcare strategy development, implementation, and execution. With a background in medical affairs and real-world evidence, she brings together differentiated study design and evidence generation solutions for value demonstration. She also has oversight of epidemiology, patient and physician services, scientific/clinical strategy, and medical writing teams.
Dr. Lytle holds a PhD in Neuroscience from Georgetown University as well as a Master of Biotechnology Enterprise & Entrepreneurship (MBEE) from Johns Hopkins University. A fellow of the American Association for the Advancement of Science (AAAS), and certified Project Management Professional (PMP), her approach is systematic and grounded in science.
The post Regulatory Submissions with Real-World Evidence: Highlights from the Joint Duke Margolis Institute and FDA Public Meeting appeared first on UBC.
]]>The post Optimizing REMS Pharmacy Integration: RAPID – UBC’s Standards-Based Solution That Minimizes Burden while Elevating Compliance and Patient Safety appeared first on UBC.
]]>Prior to product shipment, the pharmacy has a litany of steps to complete, including the added REMS verification and authorization to dispense. The current platforms and systems in place in the market require additional manual effort to perform REMS-required activities adding burden to the healthcare system.
What if that didn’t have to be the case?
As a leader in REMS Consulting, Strategy, Operations and Compliance Reporting – UBC is introducing a transformative solution that reimagines how pharmacies verify and adhere to REMS requirements, easing the burden, decreasing the number of manual processes and increases the efficiency of meeting the REMS requirements: RAPID – REMS API Data exchange.
This National Council for Prescription Drug Programs (NCPDP) standards-based, secure, application programming interface (API)-enabled service connects the Pharmacy Management System (PMS) directly to the REMS database to streamline pharmacy workflows, minimize operational burden, and elevate compliance, to help ensure patient safety.
The Current Problem: Fragmented Workflows, Manual Burden, and Compliance Risk
Today, pharmacies dispensing REMS products experience a disjointed and manual process to verify REMS requirements and report REMS-product dispenses. Pharmacy staff must exit their existing workflow to log into external REMS portals, remember multiple passwords, and manually verify stakeholder enrollment or generate REMS Dispense Authorizations (RDAs). Then, the REMS required dispense reporting often involves additional separate steps to exchange the REMS required data. These fragmented workflows introduce multiple points of failure to adhere to REMS requirements including:
- Transcription errors from manual data entry
- Missed verification steps due to human oversight
- Delays in RDA generation that slow down prescription fulfillment
- Increased non-compliance, such as invalid RDAs or unreported dispenses which can impact patient safety
This not only burdens pharmacy staff with additional time to process a REMS product but also increases the cost and risk for manufacturers and patients alike.
UBC Solution: Seamless, Standards-Based Integration. Built for Scale
Built on the NCPDP SCRIPT industry standard and adhering to HIPAA and 21 CFR requirements, RAPID ensures a secure, direct interoperability across pharmacy systems and REMS programs. It’s designed to be scalable, secure, and adaptable – capable of supporting both specialty and retail distribution models. The API operates behind the scenes, automating REMS tasks and eliminating the need for manual intervention.
This pharmacy integration allows for real-time data sharing that allows pharmacy staff to complete the following REMS activities from within their existing PMS workflow, without toggling between systems:
- Verify ETASU, such as stakeholder enrollment and certification
- Generate RDAs
- Report dispenses
Shifting from Manual to Automated
UBC’s solution is not just a technical upgrade – it’s a strategic shift in how REMS can be operationalized at the pharmacy level.
Figure 1, Illustrates differences between the current state versus the UBC-enhanced process emphasizes the importance and benefits of integration and automation:
| Traditional REMS Workflow | UBC RAPID |
| Manual portal access | Automated PMS integration |
| Multiple logins and systems | Single sign-on and workflow within PMS |
| Transcription errors | Real-time, accurate data exchange |
| Delayed RDA generation | Instant RDA response |
| Potential Non-compliance and patient risk | Built-in ETASU verification and REMS compliance |
| Additional time to process a REMS product | Reduced time to process a REMS product |
Figure 1: Current Workflow (Left), and the Revised RAPID driven Streamlined workflow (Right)
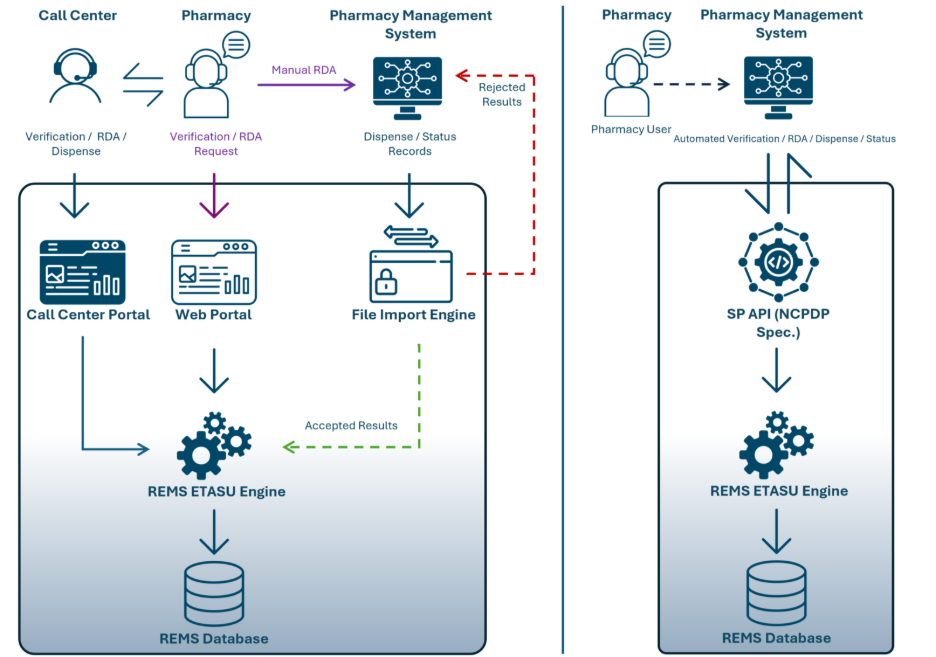
Pharmacy staff no longer need to leave their PMS or rely on memory to complete REMS tasks. Instead, verification, authorization, and reporting happen automatically, reducing errors and accelerating prescription processing.
Efficiency, Compliance, and Minimized Stakeholder Burden
UBC’s RAPID delivers measurable benefits to manufacturers:
Enhanced REMS Compliance
Invalid RDAs are a leading cause of REMS non-compliance. UBC’s integration automatically verifies the elements to assure safe use (ETASU) during the pharmacy’s regular process, reducing the risk of non-compliant dispensing and enhancing patient safety.
Industry Alignment
FDA is actively encouraging the adoption of technological innovations that integrate REMS into pharmacy workflows. UBC’s solution is built on NCPDP SCRIPT standards – ensuring compliance with FDA initiatives and positioning manufacturers as forward-thinking leaders in REMS technology.
Burden and Error Reduction
By keeping pharmacy staff within their PMS, UBC eliminates the need for external portals, reducing training requirements, time spent processing REMS products, and minimizing transcription errors. This leads to higher data quality and fewer compliance issues.
A Better Experience for Pharmacies, Manufacturers, and Patients
This isn’t just an API – it’s our strategic investment in the future of REMS and driving a better experience for those participating stakeholders.
By reducing pharmacy burden and time, improving compliance, and aligning with FDA standards, UBC’s RAPID strengthens the entire REMS ecosystem:
- Patients benefit from safer, quicker access to medications
- Pharmacies gain efficiency and clarity
- Manufacturers reduce non-compliance and risk
- FDA receives higher-quality data
UBC’s solution is already generating interest across the pharmacy network and is poised to become the new standard for REMS integration.
Let’s explore how this integration can elevate your REMS program and deliver measurable impact.
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.
About the Authors

Tracy Gentile serves as a REMS Operations Project Director at UBC. With a focus on patient safety and reducing stakeholder burden, Ms. Gentile and her team support the REMS lifecycle blending regulatory compliance, strategic planning, stakeholder engagement, and technological advancements to ensure REMS programs are compliant and successful. With almost two decades of industry experience, Ms. Gentile has worked across various therapeutic areas, indications, and risks. She has worked at UBC for the past 7 years.

Haresh Patel serves as Director of Software Engineering at UBC, where he leads the development of technology solutions that power REMS programs. He brings more than 20 years of experience in the technology industry, including over a decade dedicated to REMS.
As a solutions architect and strategic technology leader, Mr. Patel has been instrumental in shaping the technology behind the majority of UBC’s REMS programs. He partners closely with pharmaceutical manufacturers, healthcare providers, and pharmacies to deliver systems that simplify compliance, enhance patient safety, and reduce burden for all stakeholders.
The post Optimizing REMS Pharmacy Integration: RAPID – UBC’s Standards-Based Solution That Minimizes Burden while Elevating Compliance and Patient Safety appeared first on UBC.
]]>The post Is Your Real-World Data ‘Fit-for-Purpose’? The Critical Role of Feasibility Assessments appeared first on UBC.
]]>With the proliferation of available and increasingly diverse types of RWD, a critical challenge faces researchers at the onset of designing a database study: selecting the right data sources to support the research. This article provides approaches to help ensure that database feasibility assessments — conducted in advance of research to evaluate key study and logistical criteria — are successful in identifying the most relevant data sources appropriate for the evaluation of study objectives.
Identification of RWD sources with potential to satisfy evaluation criteria
Identifying a wide range of potential data sources is a crucial first step in the feasibility assessment process. Some common RWD sources include electronic medical records (EMR), patient registries, medical and pharmacy claims, and patient-generated data, such as those gathered from surveys. One of the best approaches for this first step is to review relevant published literature that present results from database or registry studies with similar objectives to the current study of interest.
Secondly, the use of online database search tools offers an efficient way to selectively identify healthcare databases that match key study criteria like geography or population demographics. These online tools include B.R.I.D.G.E. TO DATA®, an online database profiling global population healthcare databases for use in epidemiology and health outcomes, and the HMA-EMA Catalogues of RWD sources and studies. These repositories collect metadata from RWD sources and studies with the potential to help researchers identify suitable data to address specific research questions.
Importantly, researchers should consider RWD that offer unique, less traditional types of data elements not consistently found in conventional RWD sources. For instance, in the US, data from commercial patient support programs sponsored by pharmaceutical companies to improve patient access to, and overall experience with, a marketed drug may provide unique data elements for a study that is evaluating treatment patterns and non-compliance to a newly marketed product.
Establishing key criteria for selecting the appropriate data sources
An essential consideration for conducting successful database feasibility assessments is prioritizing the most important criteria for database evaluation and ultimate database selection. Publicly available documents have been issued by the FDA and the European Medicines Agency (EMA) that contain guidance to support high-quality RWE generation, with the goal of continuing to strengthen the use of RWD and associated RWE for regulatory decision-making. Two recently released documents by regulators that provide valuable insights into critical characteristics of RWD to be considered in regulatory submissions include: Journey towards a roadmap for regulatory guidance on real-world evidence (EMA, February 2025)2 and Real-world data: Assessing electronic health records and medical claims data To support regulatory decision-making for drug and biological products (FDA, July 2024).3
Some of this guidance is specific to the type of RWD, such as FDA’s recommendation that sponsors using EMRs should evaluate the completeness, accuracy, and plausibility of the data, including verifying data against the source.4 It is important to provide evidence to regulators that a selected database is appropriate for addressing the specific study question of interest. In general, the following evaluation categories should be used to assess ‘fit-for-purpose’ for selecting data sources to use in conducting the study of interest:
- Data specificity — particularly important for key variables to create relevant study cohorts and assess outcomes
- Data quality — includes data completeness, plausibility of data values, and established quality assurance/quality control plan
- Accessibility — e.g., patient-level vs aggregate data only; data must remain local to data owner vs data may leave the owner’s geographic region
- Data reliability — includes data accrual (e.g., data source and collection methods) and data assurance (e.g., processes and personnel involved in data capture to minimize errors)
Data linkage may be necessary to evaluate all study objectives
In some instances, data sources may need to be combined to provide sufficient data to address the study objectives. For example, pharmacy claims can reveal patterns in drug adherence or switching between medications, but cannot provide clinical data often needed to identify study cohorts or evaluate study outcomes. Laboratory results can be used to evaluate a number of healthcare measures, such as the impact of a drug on biomarkers, as illustrated by the potential effect of a GLP-1 therapy on hemoglobin A1c levels in patients with diabetes.5 In situations where no single data source is sufficient to generate results that address all study objectives (or may not cover all relevant timeframes required to evaluate study objectives), exploration of the potential for data linkage across patient populations using common patient identifiers (e.g., Datavant de-identified patient token) should be considered.
Evolving tools for RWD feasibility
Technological advancements are continuously improving our ability to evaluate and utilize RWD effectively. In particular, the evolution of artificial intelligence (AI) and machine learning (ML) presents numerous exciting possibilities that enhance our ability to ‘see’ into the information that is captured within a database. AI/ML enables multiple tools that can be leveraged in RWD assessment, including natural language processing, language learning models, named entity recognition, and machine vision. Natural language processing, for instance, can be used to determine if specific types of information are captured in unstructured notes in EMRs — for example, positive identification of genetic mutations as underlying etiology of a patient’s disease — which are otherwise highly challenging to obtain. Algorithms can be created to enable the efficient AI-based evaluation of databases and their suitability for studies.
Another technological advancement that enables efficient and comprehensive assessment of data captured in an RWD source (structured and unstructured) is the availability of software platforms that provide clinically validated code lists — medications, diagnoses, procedures, etc. — as a resource to researchers who are faced with evaluating data sources that differ in their underlying coding of relevant variables. Commonly referred to as Computable Operational Definitions (CODefs), these AI-informed, indication-specific libraries provide current definitions of study design elements such as disease cohort and outcome identification.
Potential implications of inadequate feasibility assessment
Importantly, serious negative consequences can result from selecting inappropriate or unreliable data to conduct a study. Failing to select suitable data sources from the outset can incur unnecessary costs by purchasing and processing datasets that do not fit research needs. In a worst case scenario, the wrong data have the potential to create bias and lead to erroneous study conclusions, damaging the researchers’ and sponsor’s reputations, and even leading to regulatory rejection.
Partnering with experts
Formal database feasibility assessments should follow a rigorous process, starting with the development of a focused data owner questionnaire to assess a wide variety of data characteristics, including population coverage, years of data availability, data elements, and potential for linkage. Documentation of rigorous methods for conducting a gap analysis to identify whether the appropriate variables needed for evaluation are present should be created and followed.
Partnering with an experienced organization such as UBC brings informed insights and years of expertise in the feasibility assessment processes. Our experienced team of data analysts, epidemiologists, clinicians, regulatory experts, and information technology specialists have conducted dozens of global database and registry feasibility assessments covering multiple data source types and across varied disease and exposed populations. Many of these assessments were to support sponsors’ regulatory requirements, such as a post authorization safety study (PASS). UBC can greatly improve a sponsor’s ability to effectively leverage RWE to support regulatory submissions and discretionary research.
Database feasibility assessments to evaluate key study and logistical criteria are a first step to ensuring the benefits of RWD/RWE are maximized for your clinical study. To learn more about how RWD/RWE can be effectively leveraged, as well as data interoperability and the generation strategies that support it, read UBC’s case study, “Enriching clinical studies with longitudinal real-world data” to better understand a specific application of RWD/RWE enrichment.
References
1. Center for Drug Evaluation and Research and Center for Biologics Evaluation and Research. (2025, June 9). Real-world evidence. U.S. Food and Drug Administration. Accessed August 11, 2025. https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence
2. Human Medicines Division/Methodology Working Party. (2025, February 19). Journey towards a roadmap for regulatory guidance on real-world evidence. European Medicines Agency. Accessed August 11, 2025. https://www.ema.europa.eu/en/documents/other/journey-towards-roadmap-regulatory-guidance-real-world-evidence_en.pdf
3. Center for Drug Evaluation and Research Center for Biologics Evaluation and Research Oncology Center of Excellence. (2024, July 25). Real-world data: Assessing electronic health records and medical claims data to support regulatory decision-making for drug and biological products. U.S. Food and Drug Administration. Accessed August 11, 2025. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/real-world-data-assessing-electronic-health-records-and-medical-claims-data-support-regulatory
4. Food and Drug Administration. “Real-World Data: Assessing Electronic Health Records and Medical Claims Data to Support Regulatory Decision-Making for Drug and Biological Products.” July 2024.
5. Tran V, Tran H, Demirel S, Thompson-Moore N. Impact of Glucagon-Like Peptide 1 Receptor Agonists in Patients with Hemoglobin A1c of 9% or Greater. J Pharm Pract. 2023;36(5):1125-1133. doi:10.1177/08971900221087933
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.
About the Authors

Irene Cosmatos, MSc, is a Senior Director of Epidemiology & Real-World Evidence at UBC. She and her team support UBC’s work that involves the use of diverse, observational healthcare databases, registries, or patient medical charts to answer sponsors’ research questions. She brings more than 25 years of experience in analyses of large, retrospective patient databases across all therapeutic areas, US and non-US, in support of epidemiologic and health outcomes research.

Jeff Lowry is the Senior Director of Technology Solutions Services at UBC leading UBC’s enterprise data services and technology solution services team. With a focus on patient identity management and study independent data model designs, Jeff and his teams have produced flexible, innovative models that integrate clinical, safety, and commercial data to support complete patient lifecycle solutions.
The post Is Your Real-World Data ‘Fit-for-Purpose’? The Critical Role of Feasibility Assessments appeared first on UBC.
]]>The post Enhancing patient engagement with technology and digital solutions appeared first on UBC.
]]>Patient engagement has a number of definitions, but can generally be understood as a patient’s attitude, active participation, and involvement in their own care. Positive quality engagement plays a vital role in patient adherence to treatment plans, in addition to supporting retention. For this reason, technologies that support engagement can be invaluable. With the advancement of technology, a variety of digital tools — including electronic health records (EHRs), electronic healthcare technology solutions, and digital assistants — have emerged with the ability to contribute meaningfully to patient engagement.
With EHR data, researchers can customize messages for outreach through EHR patient portals to distinct patient demographics, including race, gender, or age.1 Such tailored messages can make a clinical study feel more relevant to prospective participants, address the needs or concerns of specific patient groups, and build patient trust and engagement from the outset while also streamlining engagement across registries, clinical studies, and patient support services,
Electronic healthcare technology solutions
Many of the burdensome administrative tasks that might deter participation or erode patient motivation can now be managed through electronic healthcare technology solutions. eConsent tools are an excellent example, making it possible for patients to read and sign consent documents digitally. They allow patients to review the details of the clinical study at their own pace, ensuring a better understanding of the terms to which they agree, in addition to creating a smoother process.
Another way electronic healthcare solutions can benefit patient engagement is through tools, such as electronic benefit verifications and prior authorizations, which streamline insurance authorization and requirements. Navigating insurance coverage can be challenging for many patients. Technology designed to help bridge the gap between patients, and insurance providers can help ease some of the stress and confusion for patients
Digital assistants
Dedicated, AI-enabled digital assistants can help patients navigate access to therapy. They can act as guides to help set expectations about upcoming program activities, shipments, provide reminders for appointments and even help to capture patient-reported outcomes. In doing so, they help to lower many of the obstacles that patients face in staying engaged with their therapy.
By providing an easily accessible resource with 24/7 availability, digital assistants allow patients to feel better supported during their treatment journey, resulting in improved contact rates. In one instance, having access to digital assistants as a resource meant patients chose to leave therapy at an average 12% lower rate.2
Balancing technology and humanity
Technology and digital tools have a significant role to play in creating a smoother patient experience and ultimately improving outcomes. Nevertheless, there is no substitute for a human touch. Digital tool implementation should be complemented with personalized human interactions to provide an empathetic point of contact. Finding a balance between these two elements is key to truly maximizing patient engagement.
Learn more about employing technology and interpersonal connections in our white paper, High-Touch, High-Tech Patient Services for Innovative & Novel Therapies.
References
- Sherman et al. Use of patient portals to support recruitment into clinical trials and health research studies: results from studies using MyChart at one academic institution, JAMIA Open, Volume 5, Issue 4, December 2022, ooac092, https://doi.org/10.1093/jamiaopen/ooac092
- Based on UBC program data from May 2020-December 2021, including 18,000 conversations across six programs.
About UBC
United BioSource LLC (UBC) is the leading provider of evidence development solutions with expertise in uniting evidence and access. UBC helps biopharma mitigate risk, address product hurdles, and demonstrate safety, efficacy, and value under real-world conditions. UBC leads the market in providing integrated, comprehensive clinical, safety, and commercialization services and is uniquely positioned to seamlessly integrate best-in-class services throughout the lifecycle of a product.

About the Author
Dana Edwards, Vice President, Patient Access & Strategic Engagement
Dana Edwards serves as the Vice President, Patient Access & Strategic Engagement at UBC. She brings more than 20 years of experience in executing patient service and market access strategies to this role. Ms. Edwards is a strategic advisor to pharmaceutical and biotech leaders on the design and implementation of patient service programs that synchronize the right people, services, and technology for their unique patient population and therapy.
The post Enhancing patient engagement with technology and digital solutions appeared first on UBC.
]]>